Translational Perioperative and Pain Medicine (ISSN: 2330-4871)
ARTICLE DOI: 10.31480/2330-4871/065
REVIEW ARTICLE OPEN ACCESS
Signal Crosstalk Between TLR4 and Opioid Receptor Pathways
Sai Sripad Kodukula1 and Si Zeng1,2
1Department of Anesthesiology and Critical Care, Perelman School of Medicine at the University of Pennsylvania, USA
2Department of Anesthesiology, Sichuan Academy of Medical Sciences & Sichuan Provincial People's Hospital, China
Si Zeng, MD, Department of Anesthesiology, Sichuan Provincial People's Hospital, Sichuan Academy of Medical Sciences, China, E-mail: xzyxyzs@hotmail.com
Editor: Daqing Ma, MD, PhD, FRCA, Professor of Anaesthesia and BOC Chair, Head of Anaesthesia Research, Imperial College London, G3, 44 Chelsea & Westminster Hospital, 369 Fulham Road, London SW10 9NH.
Received: January 10, 2018 | Accepted: February 08, 2018 | Published: February 10, 2018
Citation: Kodukula SS, Zeng S. Signal Crosstalk Between TLR4 and Opioid Receptor Pathways. Transl Perioper & Pain Med 2018;5 (1):27-32.
Abstract
Opioid Receptors are known to share a relationship with other receptors throughout the body. However, the interaction between the mu opioid receptor (MOR) and Toll-like receptor 4 (TLR4) is of particular interest due to the observed side-effects of pain, inflammation and addiction. Here we review in detail the G-protein coupled receptors (GPCR) pathway of MOR and the kinase pathway of TLR4 and highlight the regions in these pathways where key interactions occur.These interactions are identified by analyzing the isometric differences in commonly used opioids; describing the cross-talk between GPCR signaling pathways of MOR and kinase pathways of TLR4 and linking this key relationship to inflammation and pain.
While discussing interactions, potential treatment methods such as drug cocktails and opioid recycling are highlighted in order to prevent pain and inflammation caused by the interactions.
Keywords
Opioid receptors, Toll-like receptor 4, G-protein coupled receptors, Inflammation
Introduction
Opioids have long been used to induce analgesia and relieve pain. The Toll-Like receptor 4 (TLR4) receptor is known to activate the tumor necrosis factor alpha (TNF-α) kinase pathway leading to the transcription of inflammatory cytokines and causing pain. Opioid receptors are G-protein coupled receptors (GPCR) modulating pain and many other physiological functions. However, emerging studies highlight that these opioids not only act on opioid receptors, but also interact with other receptors such as Toll-Like receptors (TLR), especially Toll-Like recpetor 4 (TLR4).
TLR4 Receptor/Pathway
TLR are found in the human body and are responsible for activating the innate immune system [1]. TLR's are known to modulate immune function which directly leads to inflammation and pain. TLR's recognize pathogen associated molecular patterns (PAMP's), which are widely expressed in pathogens such as bacteria and viruses. PAMP's activate an intracellular signaling cascade, causing the downstream activation of inflammatory cytokines which produce immune cells such as dendritic cells and glial cells, ultimately leading to inflammation and pain [2]. The TLR4 receptor specifically is most-known for the recognition of Lipopolysaccharide (LPS); a component in a gram negative bacteria. The TLR4 pathway consists of a signal transduction pathway with continuous kinase activation until the transcription of inflammatory genes in the nucleus. The process starts with LPS binding itself to the Lymphocyte antigen 96 receptor or (MD-2), associated on the surface in between TLR receptors, providing a link between LPS and the receptors [3]. The activation of the TLR4 receptor via LPS causes a conformational change which leads to the activation of downstream Toll-interleukin receptor (TIR) domains such as TIRAP, TRAM and MyD88 as shown in figure 1. Once the signal is amplified enough to deactivate the inhibitory IKK and IkBa proteins, transcription of inflammatory cytokines such as NF-kB and TNF-a occur inside the nucleus [4].
Figure 1: TLR4 Pathway: Begins with the activation of the MD2-Receptor with LPS Binding. Conformational change in TLR4 recpetor causes the downstream activation of TIRAP and Myd88 dependent pathway. Kinase-Kinase signaling to the downstream IRAK, TRAF and TAK proteins.Degradation of IKK and IKBa inhibitory factors causing the transcription of NF-kB mRNA transcripts inside the nucleus leading to the production of inflammatory cytokines.
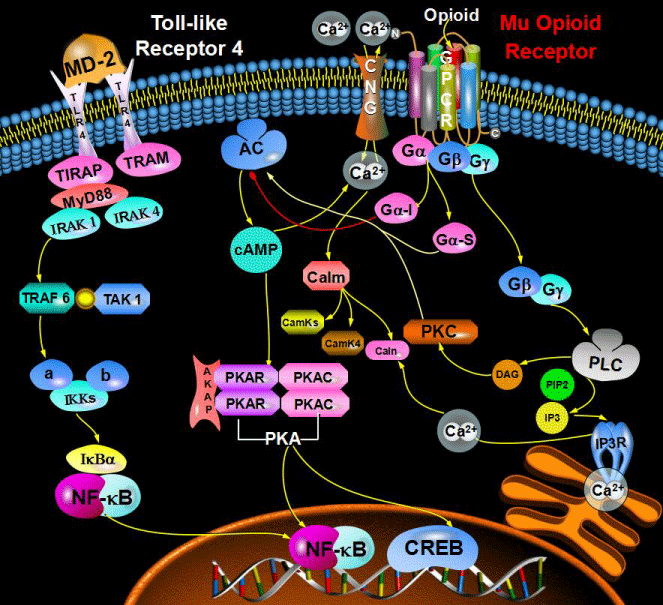
Opioid receptors are GPCR's that activate inhibitory G-proteins. These receptors form homo and hetereodimeric complexes which signal to kinase cascades and scaffold a variety of proteins, producing the desired results of analgesia and pain control [5]. Opioid recpetor signaling begins when an opioid such as morphine activates the dissociation of the Ga and GBy subunits, causing the activation of the GTPase enzyme through the modulation of sodium/potassium pumps [6,7]. GTPase enzyme controls the phosphorylation of GTP and the activation/de-activation of cAMP. Endogenous opioids stimulate GTPase activity to inhibit the production of cAMP, thus resulting in a reduced Ca2+ content in synaptic vessels causing the desired effects of analgesia, but also the undesired effects such as opiate tolerance [7-9].
There are three major opioid receptors: mu (μ, MOR), kappa (k, KOR) and delta (δ, DOR). However, the relationship between TLR4 and MOR was investigated the most. MOR activation induces analgesia, sedation and in select cases dependence and inflammation [10]. MOR activation results in the replacement of the selectively inhibiting Gβγ subunit signaling, which directly controls the downstream activation of the adenylyl cyclases [11]. Activation of adenylyl cyclases leads to the phosphorylation of other proteins, specifically PKC-1 gene, which controls the concentrations of diacylglycerol (DCG) as well as Ca2+ ions, [12] directly responsible for downstream signaling such as the activation MAP kinase proteins, ERK and p38 proteins within the nucleus which produce analgesia [13].
Opioid Receptors/Pathway
The opioid receptor (OR) system consists of KOR, MOR, and DOR, Gi/o-coupled with seven transmembrane domains. These domains are highly homological with variations occurring primarily within extracellular domains, which play an critical role in pathophysiological and physiologicalprocess [14]. Gαand Gαand Gβγ subunits of OR dissociate from one another after OR isactivated by endogenous agonist μ-opioid peptide like endorphins, or exogenous agonists likemorphine and fentanyl, subsequently acting on various intracellular effector pathways [15,16] such as adenylylcyclase (AC), cAMP, Ca2+channels, inositol triphosphate (IP 3), triglyceride (DAG) and other second messengers, protein kinases, etc. Protein phosphorylation is promoted and gene expression is altered after OR activation. The classical opioid receptor signaling is cyclic adenosine monophosphate (cAMP) - protein kinase (PKA) pathway, which activates pertussis toxin sensitive G protein (Gi), inhibits adenylylcyclase activity and intracellular cAMP production. CAMP as the second messenger for this pathway can regulate the pathway by activating PKA [17]. Intracellular Ca2+ level is reduced by inhibiting the activity of calcium channel via Ca2+ dependent protein kinase (PKC) pathway. Calmodulin also plays a vital role in the signal transduction, once the second messenger is activated and the transcription factors and regulatory factors are modified.
Relationship of TLR4 to Opioid Receptors
Enantiomers/Stereo-selectivity
Recent experiments conducted on understanding the relationship between TLR4 and Opioid Receptors highlighted the importance of stereo-selectivity of opioid ligands. In several knockout studies it was found that various enantiomers/ uncommon isomers of drugs such as morphine and naloxone were found to activate different signal transduction pathways. Unnatural enantiomers of opioid agonists such as (+)-morphine were found to activate TLR4, but lack opioid receptor activation [18]. While, unnatural enantiomers of opioid antagonists such as (+)-naloxone were found to specifically inhibit TLR4, while lacking MOR affinity [19]. Thus, when testing various opioid receptor agonists such as morphine and oxycodone, the stereo-selectivity of TLR4 and the opioid receptors play an important role in the effectiveness of the drug. When morphine is administered, the (-) Morphine enantiomer acts as a mu opioid agonist and leads to analgesia, however, the (+) morphine enantiomer activates TLR4, which leads to the activation of TNF-a and IL-1, leading to the attenuation of analgesia. This attenuation of analgesia leads to a higher requirement of morphine administration leading to tolerance and addiction [20,21].
Crosstalk between GPCR and Kinase Pathways
LPS induced activation of TLR 4 leads to the observed effects such as inflammation or analgesia. In the past decade research has focused on the "crosstalk" that might occur between the GPCR pathway in opioid receptors and the TLR 4 pathway. In MOR expressed in dendritic cells it was observed that a small dosage of MOR agonist: morphine led to the inhibition of TLR4, specifically the nF-KB pathway, which directly contributed to analgesia. However, when a high dosage of morphine was administered, the contrary was observed; ThenF-KB mRNA were found in high concentrations within the nucleus hinting at a cross-talk between the GPCR's and Kinases. Since, nF-KB is produced through the TLR4 pathway as shown in figure 1, a cross-talk had to have occurred between the GPCR activated by the morphine and the TLR4 Kinase pathway, which lead to the transcription of nF-KB and ultimately the production of more dendritic cells [22,23].
A few speculations on the cross-talk involves the idea of varying isomers described above, where chronic amounts of morphine could contain a mixture of (+) and (-) morphine, causing the activation of TLR4 receptors [20,21]. Another mechanism for cross-talk could involve the use of downstream effector pathways that are activated by GPCR's. Downstream effector pathways include the cAMP/PKA/CREB in GPCR's and PLC/PKC/IKK in Kinase pathways [24]. Hence, the activation of cAMP or any of the intermediate domains within the GPCR pathway could trigger a signal in the kinase pathway activating IKK and ultimately leading to the transcription of nF-KB and other inflammatory factors [25].
Crosstalk Effects on Pain and Inflammation
The interaction between TLR4 and MOR share a direct effect on pain and inflammation. Allodynia and hyperalgesia are direct consequences of inflammation caused by the activation of downstream TLR4 kinases by MOR. TLR4 mediated TNF-a activation leads to the production of HMGB-1 protein which triggers the transcription of macrophages and dendritic cells. These immune cells attenuate the effects of opioids as well as cause excessive neuronal activity causing heightened neuropathic pain [22,26]. As a result of TLR4 activation, a higher dosage of opioids is being used to achieve the same result leading to dependence and addiction. While analyzing opioid dependence, the type of opioids used during procedures were also scrutinized for their effects on pain and inflammation. Morphine metabolized into two active metabolites: metabolite morphine-3-glucuronide (M3G) and morphine-6-glucuronide (M6G).In a regular morphine dose, a metabolite M3G was shown to play a significant role in inflammation/pain, as it activates TLR4 and opposes morphine analgesia causing agitation and hyperalgesia within knockout mice [27-29]. In contrast, another metabolite M6G binds to MOR and acts as a potent analgesic.In order to control the pain and inflammation induced by TLR4 activation, a variety of opioid cocktails have been tested. Buprenorphine (a MOR agonist) combined with a dose of LPS-ultrapure (TLR4 antagonist) was measured for its effectiveness. Results in multiple knock-out experiments showed a higher analgesia and lower inflammation/overall pain . Thus, select TLR4 antagonists such as LPS-ultrapure and naloxone (+) could be targets for pain relief and drug rotation in order to prevent dependence [30,31].
TLR4 and opioid receptors are found in immune cells located primarily in our thalamus, spine and blood stream. Recent studies have linked the severity and duration of pain and inflammation to be based on the location of the TLR4 receptors. Morphine binds to the neuronal μ-opioid receptor and the innate immune TLR4 localized primarily on microglia. Neuroinflammatory effects are initiated which opposes the analgesic effects of morphine. It is reported that TLR4 inhibition in the periaqueductal gray (PAG) of females reverses the sex differences, which may call for the necessity for sex-specific research and promote individualized treatment strategies for opioid-based pain management.in men and women [32]. In a sample of dendritic cells taken from the spinal cord of knock-out mice, inflammation was shown to be gender dependent, where the activation of TLR4 brought forth Allodynia and hyperalgesia in male mice, but not female ones. After a thorough analysis the cause for this disparity was shown to be testosterone dependent. Due to the high testosterone content in male mice, a higher inflammatory response was noted in the spinal cord TLR4 receptors [33,34]. The following observation only held significance in the cell samples from the spinal cord. In different sample of glial cells in the brain, TLR4 activation by opioid receptor crosstalk was shown to activate a downstream kinase Iinterluekin-1 (IL-1), responsible for immune regulation, specifically IL-a [35]. Drugs such as morphine and adrenorphine are IL-1 agonists, resulting in TLR4 activation leading to a high amount of neuropathic pain [36]. Mast cells with sickle cell anemia were another such cells with interesting data on pain and inflammation after the activation of their TLR4 receptors. In a sample of normal sickle mast cells, often times a lack of proper MOR activation caused by a defective HbSS-BERK protein, which normally inhibits MOR crosstalk, caused a higher amount pain. However, while observing the activity of TLR4 knockout mice with sickle-cell anemia, overall neuropathic pain after the administration of opioids was significantly lower. Thus, the experiment linked the TLR4 receptor to neurogenic inflammation and hyperalgesia in sickle mice [37]. The cross-talk between GPCR's and TLR4 has a direct impact on pain and inflammation and drugs specifically acting as TLR4 antagonists may help to reduce neuropathic pain and immune responses to opioids.
Conclusion
The relationship between MOR and TLR4 is of great significance, as inflammation, pain and dependence are considered one of the biggest drawbacks of opioid usage. The observed cross-talk between MOR and TLR4 causes the activation of downstream kinases in the TLR4 pathway resulting in the transcription of inflammatory cytokines causing inflammation and pain. However, the degree of pain and inflammation can vary depending on the isometric differences in commonly used opioids; location of the MOR and TLR4 receptors as well as the dosage of opioids administered. In order to control inflammation a few methods such as utilizing the stereo-selectivity of opioids and selecting a cocktail of TLR4 antagonists and MOR agonists such as buprenorphine or naloxone (+) can help to prevent inflammation and prevent pain [38]. Another method could include to further explore topics of interest in this field such as the effects of kappa and delta opioid agonists on TLR4, which could potentially cause inflammation. Along with the effects of KOR and DOR agonists, the activation of other toll like receptors such as TLR2 by opioids could also be explored as recent studies have already linked TLR2 as well as TLR4 to immune responses. Thus, addressing these issues associated with TLR4 and MOR crosstalk can help to prevent unwanted inflammation as well as pain.
References
- Rock FL, Hardiman G, Timans JC, et al. A family of human receptors structurally related to Drosophila Toll. ProcNatlAcadSci U S A. 1998;95(2):588-593.
- Takeuchi O, Akira S. Toll-like receptors; their physiological role and signal transduction system. IntImmunopharmacol. 2001;1(4):625-635.
- Sachiko Akashi YN, Hirotaka Ogata, Masato Oikawa, et al. Human MD-2 confers on mouse Toll-like receptor 4 species-specific lipopolysaccharide recognition. Oxford Journals: Internal Immunology. 2001;13(12):1595-1599.
- Chow JC, Young DW, Golenbock DT, et al. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J Biol Chem. 1999;274(16):10689-10692.
- Al-Hasani R, Bruchas MR. Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology.2011;115(6):1363-1381.
- Terenius L. Stereospecific interaction between narcotic analgesics and a synaptic plasm a membrane fraction of rat cerebral cortex. ActaPharmacolToxicol.1973;32(3):317-320.
- Caruso TP, Takemoi AE, Larson DL, et al.Chloroxymorphamine, and opioid receptor site-directed alkylating agent having narcotic agonist activity.Science.1979;204(4390):316-318.
- Lord JA,Waterfield AA, Hughes J, et al. Endogenous opioid peptides: multiple agonists and receptors. Nature.1977;267(5611):495-499.
- Lemke TLW, David H.; Foye, William O. .Opioid Analgesics; Fries, DS.Foye's principles of medicinal chemistry.2002.
- Liu XY, Liu ZC, Sun YG, et al. Unidirectional cross-activation of GRPR by MOR1D uncouples itch and analgesia induced by opioids. Cell.2011;147(2):447-458.
- Mathews JL, Smrcka AV, Bidlack JM. A novel Gbetagamma-subunit inhibitor selectively modulates mu-opioid-dependent antinociception and attenuates acute morphine-induced antinociceptive tolerance and dependence. J Neurosci.2008;28(47):12183-12189.
- Ali ES,HuaJ, Wilson CH, et al. The glucagon-like peptide-1 analogue exendin-4 reverses impaired intracellular Ca(2+)signalling in steatotic hepatocytes. BiochimBiophysActa.2016;1863(9):2135-2146.
- Russo SJ, Bolanos CA, Theobald DE, et al. IRS2-Akt pathway in midbrain dopamine neurons regulates behavioral and cellular responses to opiates. Nat Neurosci.2007;10(1):93-99.
- Pradhan AA, Smith ML, Kieffer BL, et al. Ligand-directed signalling within the opioid receptor family. Br J Pharmacol. 2012;167(5):960-969.
- Childers SR, Snyder SH. Guanine nucleotides differentiate agonist and antagonist interactions with opiate receptors. Life Sci. 1978;23(7):759-761.
- Childers SR, Creese I, Snowman AM, et al. Opiate receptor binding affected differentially by opiates and opioid peptides. Eur J Pharmacol. 1979;55(1):11-18.
- Ream Al-Hasani, Michael R. Bruchas. Molecular Mechanisms of Opioid Receptor-Dependent Signaling and Behavior.Anesthesiology. 2011; 115(6):1363-1381.
- Shen CH, Tsai RY, Shih MS, et al. Etanercept restores the antinociceptive effect of morphine and suppresses spinal neuroinflammation in morphine-tolerant rats. AnesthAnalg. 2011;112(2):454-459.
- Wu HE, Hong JS, Tseng LF.Stereoselective action of (+)-morphine over (-)-morphine in attenuating the (-)-morphine-produced antinociception via the naloxone-sensitive sigma receptor in the mouse. Eur J Pharmacol. 2007;571(2-3):145-151.
- Shavit Y, Wolf G, Goshen I, et al. Interleukin-1 antagonizes morphine analgesia and underlies morphine tolerance. Pain.2005;115(1-2):50-59.
- Mohan S, Davis RL, DeSilva U, et al. Dual regulation of mu opioid receptors in SK-N-SHneuroblastoma cells by morphine and interleukin-1β: evidencefor opioid-immune crosstalk. JNeuroimmunology. 2010;227(1-2):26-34.
- Yang H, Hreggvidsdottir HS, Palmblad K, et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. ProcNatlAcadSci U S A. 2010;107(26):11942-11947.
- Shafie A, Moradi F, Izadpanah E, et al. Neuroprotection of donepezil against morphine-induced apoptosis is mediated through Toll-like receptors. Eur J Pharmacol.2015;764:292-297.
- Chen YL, Law PY, Loh HH. The other side of the Opioid story: modulation of cell growth and survival signaling. Curr Med Chem. 2008;15(8):772-778.
- Grace PM, Maier SF, Watkins LR. Opioid-induced central immune signaling: implications for opioid analgesia. Headache.2015;55(4):475-489.
- Yang H, Tracey KJ. Targeting HMGB1 in inflammation.BiochimBiophysActa.2010;1799(1-2):149-156.
- Bertram G. Katzung SBM, Anthony J. Trevor. Basic & Clinical Pharmacology. 2012;12th.
- Roeckel LA, Le Coz GM, Gaveriaux-Ruff C, et al. Opioid-induced hyperalgesia: Cellular and molecular mechanisms. Neuroscience.2016;338:160-182.
- Farzi A, Halicka J, Mayerhofer R, et al. Toll-like receptor 4 contributes to the inhibitory effect of morphine on colonic motility in vitro and in vivo. Sci Rep. 2015;5:9499.
- Agnieszka M. Jurga,EwelinaRojewska, Anna Piotrowska, et al. Blockade of Toll-Like Receptors (TLR2, TLR4) Attenuates Pain and Potentiates Buprenorphine Analgesia in a Rat Neuropathic Pain Model. Neural Plast.2016;2016:1-12.
- Eriksen GS, Andersen JM, Boix F, et al. Comparison of (+)- and (-)-Naloxone on the Acute Psychomotor-Stimulating Effects of Heroin, 6-Acetylmorphine, and Morphine in Mice. J PharmacolExpTher.2016;358(2):209-215.
- Doyle HH, Eidson LN, Sinkiewicz DM, et al. Sex Differences in Microglia Activity within the Periaqueductal Gray of the Rat: A Potential Mechanism Driving the Dimorphic Effects of Morphine. J Neurosci. 2017;37(12):3202-3214.
- Sorge RE, LaCroix-Fralish ML, Tuttle AH, et al. Spinal cord Toll-like receptor 4 mediates inflammatory and neuropathic hypersensitivity in male but not female mice. J Neurosci.2011;31(43):15450-15454.
- Eidson LN, Inoue K, Young LJ, et al. Toll-Like Receptor 4 Mediates Morphine-Induced Neuroinflammation and Tolerance via Soluble Tumor Necrosis Factor Signaling. Neuropsychopharmacology.2017;42(3):661-670.
- Brosnahan AJ, Jones BJ, Dvorak CM, et al. Morphine attenuates apically-directed cytokine secretion from intestinal epithelial cells in response to enteric pathogens. Pathogens.2014;3(2):249-257.
- Igor L. Calil, Ana C. Zarpelon, Ana T.G. Guerrero, et al. Lipopolysaccharide Induces Inflammatory Hyperalgesia Triggering a TLR4/MyD88-Dependent Cytokine Cascade in the Mice Paw. PLOS One.2014;9(3):e9013.
- Derek Vang, Rocio D Saavedra Pena, Sonia A Robiner, et al. Toll-Like Receptor 4 Knockout Attenuates Neurogenic Inflammation and HyperalgesiaIn Sickle Mice. Blood.2013;122:732-732.
- Bihel F. Opioid adjuvant strategy: improving opioid effectiveness. Future Med Chem. 2016;8(3):339-354.